Theory and Defination :
When treated with a strong base such as sodium ethoxide, two molecules of a carboxylic ester with two α hydrogen combine to give a β-keto ester in a reaction called the Claisen condensation.
It seen that Claisen condensation of esters is very similar to aldol condensation .The enolate form of one ester molecule carries out nucleophilic attack on the carbonyl carbon of another ester molecule.
How Claisen condensation differs from aldol condensation illustrates a general difference in the reactivity of esters vs. aldehyde and ketone.
In Claisen condensation, the enolate form of one ester molecule approaches another, similarly to aldol condensation, but, in this case, the tetrahedral intermediate resolves itself along an acyl substitution pathway. Both the aldol and Claisen condensations begin with an α-substitution, but in aldol condensation the overall pathway corresponds to nucleophilic addition, while Claisen condensation resolves itself in the manner of an acyl substitution reaction with sp2-hybridization returning with the departure of the leaving group.
General Reaction with illustration :

The most commonly used strong base in organic reactions, hydroxide ion, is not suitable for Claisen condensation because it could cause saponification of the ester. The base of choice in Claisen condensation is the alkoxide ion corresponding to the alkoxy group in the ester. Other alkoxides could cause trans-esterification of the ester. Since the β-ketoester formed in Claisen condensation is converted to the corresponding enolate ion by the base, in order to isolate the β-ketoester, when the reaction is complete, the reaction mixture needs to be acidified.
Mechanism:
Step 1: The alkoxide ion deprotonates the enolizable ester reversibly.


Step 4: The alkoxide ion deprotonates the β-ketoester irreversibly.

Step 5: The acid protonates enolate ion 2.

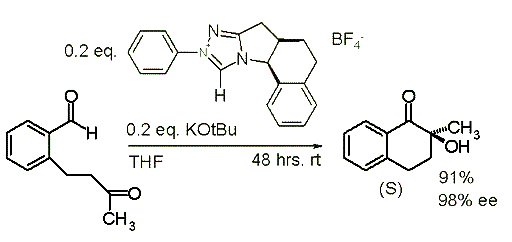
Example and Application :
1)an intra molecular rearrangement










 ;
;  , then the rate equation is:
, then the rate equation is:![\frac{d[A]}{dt} = -k_1 [A]](https://lh3.googleusercontent.com/blogger_img_proxy/AEn0k_v6Z_TJ8ttY_caCNqbnMfsDINbD-N5gILJsjx-IN2inLiF_hGUGLU36m6LNWlVijXcx6UfaJMzoiAtOXIpp423-Nr1mMsdco9y2EUn2tY7mFCHU4g3hpj5BderAsfWZDm70xqaljspScBZq4sPPjQ=s0-d)
![\frac{d[B]}{dt} = k_1 [A] - k_2 [B]](https://lh3.googleusercontent.com/blogger_img_proxy/AEn0k_s6A1W3Y9Q_qFa8ZF62zHgGUCtmnAsz5LeBiwMn3FMGolTzGyn9dEmn7lDQAkKlYebQcncxXZFJ_NXApr4yFlolcCiFm4juoj3iyu3YZsk0UrhSk5vqcacvZwJPSGKo-9qi9UlRQYYeizEa5A4eGw=s0-d)
![\frac{d[C]}{dt} = k_2 [B]](https://lh3.googleusercontent.com/blogger_img_proxy/AEn0k_tFXlD2umzSaWBwNOEenAg-QaCflCgMLrxsr9I-9ol2DS6HY2_KmR3TuQy9rAjK1Qp4yDDZA6jIem-rYgUfp0okZXVnVkJMaeVVIvcAAkUmyBlv-VQ21J0GiAoFz3wF4pvzUPFQifwz4r3oq8dvZQ=s0-d)